
Aree di intervento » Cura delle patologie dell’Ipofisi
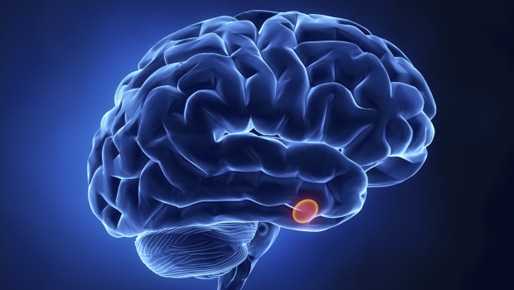 L’ipofisi è una piccola ghiandola di circa 15 mm situata praticamente al centro della base cranica ed è costituita da due lobi, uno anteriore (l’adenoipofisi), l’altro posteriore (la neuroipofisi). La si può considerare una sorta di “centralina” che governa tutte le ghiangole endocrine ed altri tessuti distribuiti nel corpo umano, influenzando la tiroide (attraverso il TSH), le gonadi (attraverso FSH e LH), il surrene (attravero l’ACTH), la mammella (attraverso la PRL), il fegato, i muscoli, i reni (attraverso il GH).
L’ipofisi è una piccola ghiandola di circa 15 mm situata praticamente al centro della base cranica ed è costituita da due lobi, uno anteriore (l’adenoipofisi), l’altro posteriore (la neuroipofisi). La si può considerare una sorta di “centralina” che governa tutte le ghiangole endocrine ed altri tessuti distribuiti nel corpo umano, influenzando la tiroide (attraverso il TSH), le gonadi (attraverso FSH e LH), il surrene (attravero l’ACTH), la mammella (attraverso la PRL), il fegato, i muscoli, i reni (attraverso il GH).
MALATTIE DELL’IPOFISI
L’ipopituitarismo è la sindrome caratterizzata dalla ridotta o assente secrezione di uno o più ormoni ipofisari con conseguente malfunzionamento delle ghiandole periferiche corrispondenti; la carenza di tutti gli ormoni ipofisari è detto pan-ipopituitarismo, il deficit di due o più ormoni ipopituitarismo parziale, mentre il deficit di un solo ormone viene definito ipopituitarismo selettivo. Le cause dell’ipopituitarismo possono essere molteplici, ma le più comuni, in età adulta, sono sicuramente i tumori ipofisari (adenomi ipofisari) e la chirurgia ipofisari eseguita per l’asportazione di un tumore (causa iatrogena). Nel bambino sono più frequenti i tumori ipotalamici che determinano compressione ipofisaria (come il craniofaringioma) e le forme congenite. Le cosiddette “cause funzionali” che possono determinare un cattivo funzionamento transitorio dell’ipofisi sono l’anoressia nervosa, la malnutrizione, la depressione.
Una delle cause più frequenti di consulto endocrinologico per un problema ipofisario è l’iperprolattinemia, cioè l’aumento dei livelli di prolattina nel sangue, che può essere causa nelle donne di alterazioni del ciclo, galattorrea (secrezione spotanea o provocata di liquido simile al latte dal capezzolo) ed infertilità. Dopo attenta anamnesi che avrà escluso le cause fisiologiche e farmacologiche (molti farmaci, anche di uso comune, possono indurre iperprolattinemia), si dovrà prendere in considerazione la possibile presenza di adenomi ipofisari prolattina-secernenti od altre patologie non ipofisarie quali l’ipotiroidismo, l’insufficienza renale ed epatica, la sindrome dell’ovaio policistico.
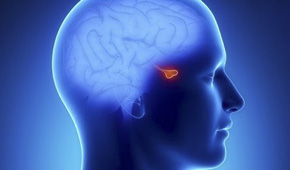 Gli adenomi ipofisari sono neoplasie benigne che originano dalle cellule epiteliali dell’adenoipofisi. Gli adenomi “funzionanti” sono quelli che producono in maniera eccessiva un dato ormone; quelli più frequenti sono i prolattina-secernenti (40-50%), seguiti da quelli che producono GH (20-25%), poi quelli che producono ACTH (8-10%); rarissimi quelli producenti TSH, FSH ed LH. I cosiddetti “macroadenomi”, cioè quelli che hanno tendenza a crescere ed espandersi comprimendo quindi le strutture cerebrali vicine di solito si presentano clinicamente con sintomi legati all’”effetto massa” quali cefalea intensa, disturbi visivi, disturbi neurologici. Come detto i più frequenti adenomi ipofisari sono i prolattinomi che fortunatamente tendono a presentarsi come microadenomi (di grandezza inferiore al centimetro) e di solito non crescono; sono responsabili dei segni e sintomi legati all’iperprolattinemia e di solito vengono trattati con sola terapia medica. Discorso diverso per i tumori secernenti GH (che danno acromegalia, con progressiva aumento delle estremità, soprattutto mani e piedi, e vistose alterazioni del volto) e quelli secernenti ACTH (che determinano la cosidetta Malattia di Cushing, con iperproduzione di cortisolo): essi si presentano di solito come macroadenomi e vengono spesso inviati al neurochirurgo.
Gli adenomi ipofisari sono neoplasie benigne che originano dalle cellule epiteliali dell’adenoipofisi. Gli adenomi “funzionanti” sono quelli che producono in maniera eccessiva un dato ormone; quelli più frequenti sono i prolattina-secernenti (40-50%), seguiti da quelli che producono GH (20-25%), poi quelli che producono ACTH (8-10%); rarissimi quelli producenti TSH, FSH ed LH. I cosiddetti “macroadenomi”, cioè quelli che hanno tendenza a crescere ed espandersi comprimendo quindi le strutture cerebrali vicine di solito si presentano clinicamente con sintomi legati all’”effetto massa” quali cefalea intensa, disturbi visivi, disturbi neurologici. Come detto i più frequenti adenomi ipofisari sono i prolattinomi che fortunatamente tendono a presentarsi come microadenomi (di grandezza inferiore al centimetro) e di solito non crescono; sono responsabili dei segni e sintomi legati all’iperprolattinemia e di solito vengono trattati con sola terapia medica. Discorso diverso per i tumori secernenti GH (che danno acromegalia, con progressiva aumento delle estremità, soprattutto mani e piedi, e vistose alterazioni del volto) e quelli secernenti ACTH (che determinano la cosidetta Malattia di Cushing, con iperproduzione di cortisolo): essi si presentano di solito come macroadenomi e vengono spesso inviati al neurochirurgo.
Il diabete insipido è invece la più frequente malattia che interessa la neuroipofisi; è determinato di solito da una ridotta produzione di ADH, ormone antidiuretico, e si caratterizza clinicamente per l’incapacità del soggetto a concentrare le urine. Ciò determina poliuria (il paziente urina più di 2,5 litri al giorno potendo arrivare fino ai 10-15 litri) e per compensare tale meccanismo è costretto a bere continuamente. Le cause possono essere relative ad un danno ipotalamo-ipofisario (tumori ipotalamo-ipofisari), ad un trauma cranico pregresso, ma spesso la causa rimane ignota (forma idiopatica). La terapia è medica, sostituendo il più delle volte l’ormone mancante (l’ADH).